ncHMR detector
Overview
Histone modification regulators (HMR) play important roles in many biological processes and function by catalyzing or binding known histone modifications. Abundant studies mapped the genome-wide profiles of HMRs through ChIP-Seq but most of them only focused on the relationship between HMRs and their known histone modification substrates/products. However, there were still some studies showed that several HMRs can bind to non-classical sites (defined as without colocalization of known histone modifications) which were involved in development, differentiation et al. Thus, we developed ncHMR detector, a computational framework to systematically predict non-classical functions and cofactors of a given HMR.
We developed software and webserver for users to utilize our framework. The usage of webserver is introducted below.
Input files
1. ChIP-seq peak file (required)
The input ChIP-seq peak file is a tab-delimited plain-text file in Browser Extensible Data (BED) format. The genome version of peak file should be hg38 or mm10. This type of file could be generated from MACS2 ChIP-seq analysis software. The BED format plain-text file has at least the following 3 tab-separated columns.
- chromosome
- chromStart
- chromEnd
Please note that the line number of bed files should be 1000~10000. Users have ChIP-seq bed file with more than 10000 peaks can rank peaks and select top 10000 peaks or use our software. We provide an example file for users to learn the usage of webserver.
Arguments
1. Cell type
ncHMR detector supports 6 ChIP-seq data-rich cell types (GM12878, K562, HeLa, HepG2, hESC, mESC) now in human and mouse. User must select the correct cell type for your HMR because ncHMR detector will scan cofactor candidates using ChIP-seq data from selected cell type.
2. HMR name
You can set a name for your HMR and the name will be showed in the output webpage as mentioned below.
3. HM substrates/products
ncHMR detector relies on significantly enriched co-occurrence of cofactor binding events and the absence of classical histone modification (HM) substrates/products. We provide qualified ChIP-seq data for available HM substrates/products in 6 cell types, at least one substrate/product must be selected. We provide two additional options for users to select multiple substrates/products simultaneously as some HMRs catalyze or bind multiple substrates/products.
4. Extended peak width (+/-)
It has been reported that the widths of binding region of distinct histone modifications are different. For instance, H3K9me3, H3K27me3 and H3K36me3 have broader binding regions. So we provide an option for users to choose the window width around HMR's ChIP-seq peak centers to calculate the signals of histone modifications. The default setting is 5kb (i.e. upstream and downstream 5kb flanking each HMR's peak centers), which might be appropriate for broad histone modifications.
5. R-squared cutoff
In ncHMR detector, we used an adjusted R-squared to evaluate the negative correlation between cobinding occurrence of cofactors and histone modification signals at HMR ChIP-seq peaks. Factors showed higher R-squared than the threshold were regarded as cofactors of non-classical functions. If none of factors show stronger negative correlation than the R-squared threshold, ncHMR detector will report that it can't detect any non-classical functions. Higher R-squared cutoff means less non-classical functions, so you can choose lower R-squared cutoff to try again when you can't obtain any predictions with high R-squared cutoff. The webserver provides an option for users to set the cutoff and the default setting is 0.1. Please note that the value should be 0~1.
6. P-value cutoff
In ncHMR detector, we permuted the cobinding events between factors and the given HMR for 1,000 times to test the significance of the calculated correlation coefficient. The webserver provides an option for users to set the p-value for statistical significance. Please note that the value shouldn't be less than 0.001 because of limited permutation experiment times.
Output
1. Summary page
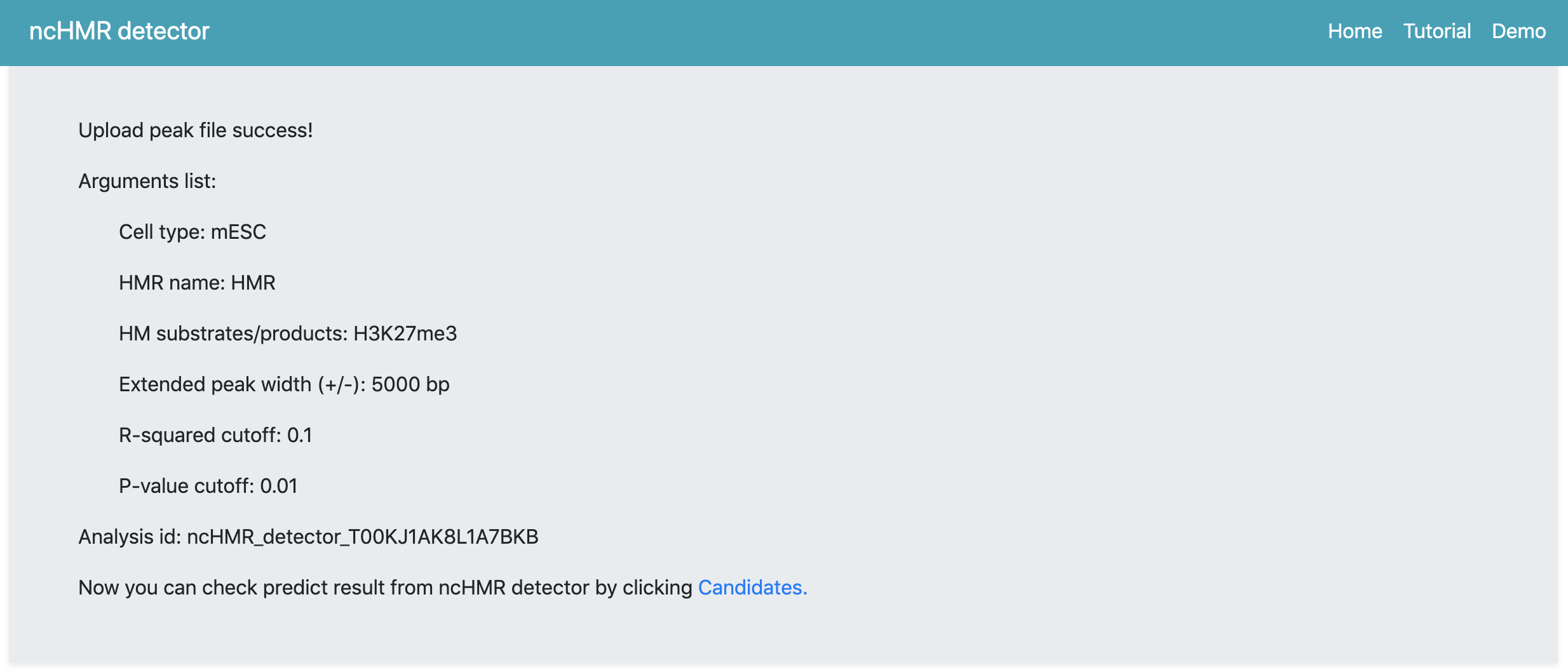
After submisson, the webserver will begin the anlysis automatically and then the webpage will be updated to show a summary page. The summary page (Figure 1) will show arguments you set to run ncHMR detector and the link pointing to the webpage containing prediction result. The webserver assigns a random analysis id for each request.
2. Candidates page
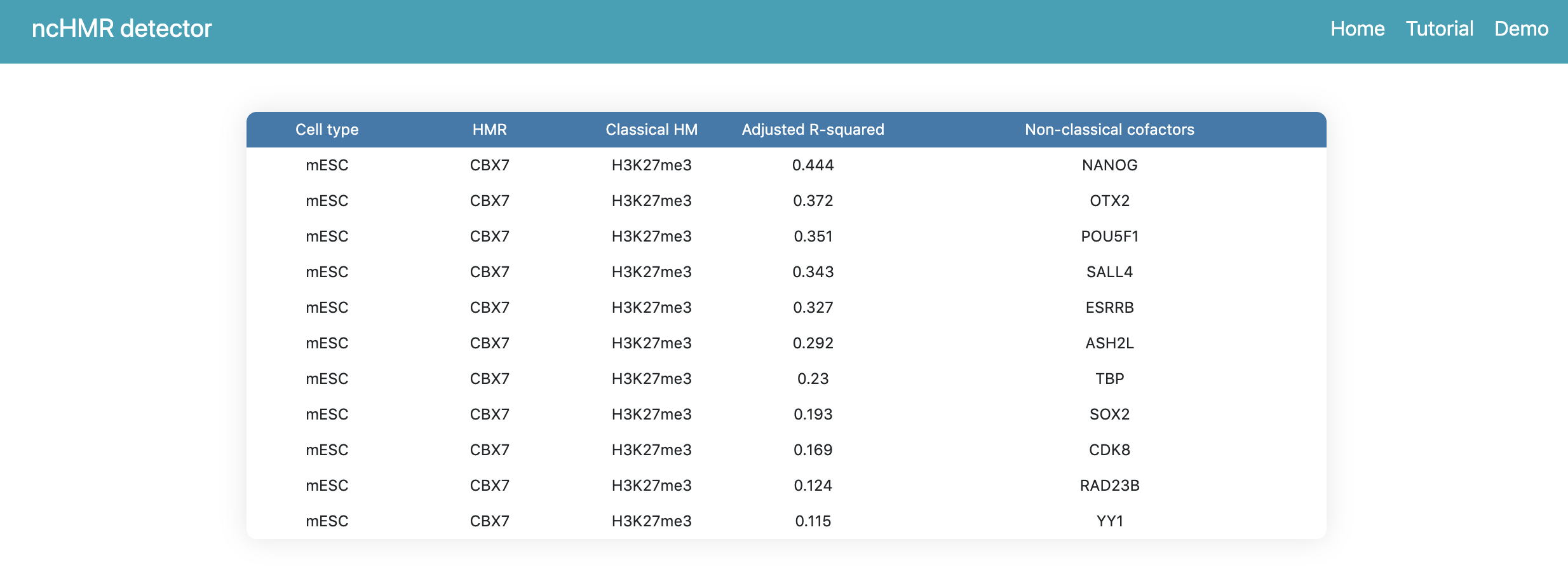
If users click the link in the summary page, the webserver will return a webpage to show the prediction result like Figure 2, the table contains all cofactors ranked by R-squared.
Contact
If you have any question abuout the usage of webserver, please contact us at tj_zhanglab {at} outlook {dot} com